


How to Eliminate the qPCR underestimation
Calling and challenging for quantitatively eliminating the underestimation of the DNA contamination in the qPCR


◆The estimated amount of the DNA contamination when the underestimation is eliminated significantly exceeds the regulation amount.
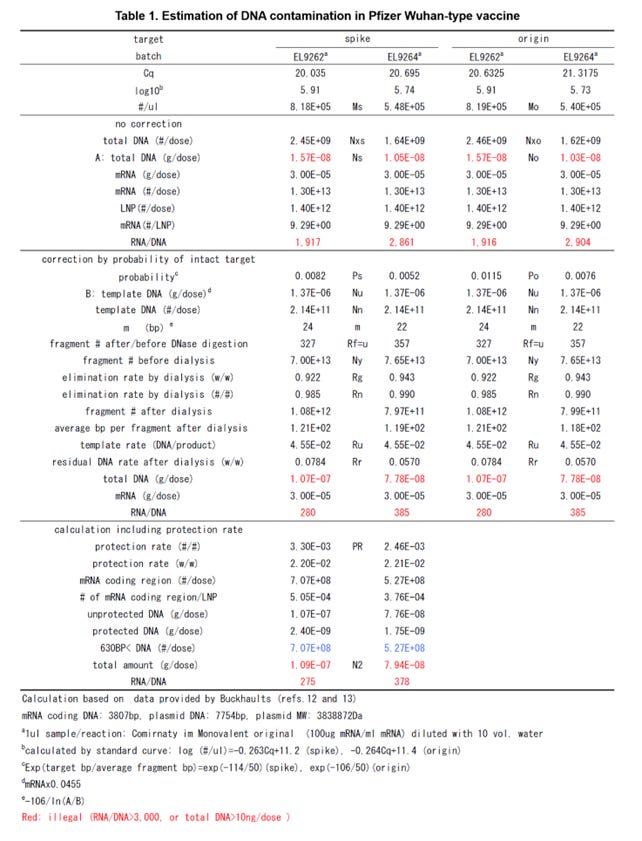
Regarding the Pfizer Wuhan-type vaccine according to the study of Buckhaults, the estimated amount of the DNA contamination of the lot number EL9262 is 109 [ng/dose], and the estimated amount of the DNA contamination of the lot number EL9264 is 79.4 [ng/dose]. Additionally, regarding the Moderna vaccine according to the study of Mckernan, the analogical estimated amount of the DNA contamination is 116 [ng/dose].
The problem of the plasmid DNA contaminations in the Covid mRNA vaccines was reported by McKernan in the substack on February 16, 2023, and the preprint thereof was filed on April 10 in the same year (ref. 1). In this preprint, three DNA contamination amounts were identified by means of three methods including the qPCR. It is known that the qPCR underestimates the DNA contamination amount due to natures of the mRNA vaccine manufacturing method and the measurement method.
For example, Moderna has self-acknowledged the underestimation of residual DNA (DNA contamination) in the qPCR in their patent US10077439B2 (ref. 2, p.16, section 19), and this discovery is still fresh in our minds. The filing date of this patent is March 13, 2014, before the outbreak of the new coronavirus, and one of the inventors of this patent is Stephane Bancel, the CEO of Moderna. Moderna also has explicitly self-acknowledged in this patent that the residual DNA is to be oncogenic and must be removed (p.7, section 1).
Furthermore, another Moderna patent US13/791922 (US20130259924A1, Stephane Bancel, et al.) has been incorporated into the above patent (ref. 2, p.10, section 8) by reference for all purpose teaches that the DNA introduced into a cell can be inherited by offspring (ref. 3, para [0006]). Additionally, the other Moderna patent US20190240317A1 teaches the insertional mutagenesis due to the naked plasmid DNA (ref. 4, para [0012]).
The above patent US10077439B2 (ref. 2) has cited the FDA guideline (ref. 5) in its end of the specification (p.18, section 24), and this FDA guideline teaches multiple risks related to the residual DNA including insertional mutagenesis, oncogenesis, and genetic integration (ref. 5, p.37).
It is obvious from those facts that the DNA contamination has a different mechanism of action from an originally intended mechanism of action of the vaccine, and it goes without saying that the DNA contamination may cause a fatal side effect on the human body.
Despite Moderna and FDA were aware of the above events, Moderna used the qPCR to detect the residual DNA amount in their actual manufacturing process for the mRNA vaccine (ref. 6, p.109), and FDA permitted it. They have continued to ignore the problem of residual DNA to this day even though they have been aware of the risks of residual DNA.
FDA has set the regulation amount of 10 [ng/dose] and below 200 [bp] for the DNA contamination (ref. 5, p.37). However, this is not the regulation amount for the residual DNA wrapped by the LNP (LNP-DNA), but for the naked DNA.
The LNP-DNA has superior long-term durability to the naked DNA and has the property of easily inducing the residual DNA into a cell. It is therefore obvious that the LNP-DNA has a different mechanism of action from that of the naked DNA, and it is extremely inappropriate for the previous regulation amount (=10 [ng/dose], <200 [bp]) to be directly applied to the new LNP-DNA technology.
However, since there is no regulation amount for the LNP-DNA, we cannot help but following the previous regulation amount for the naked DNA. An act of wrapping the residual DNA with the LNP and introducing it into the human body is an ethical issue and should not be permitted in any amount since safety cannot be guaranteed.
It goes without saying that their such acts were caused intentionally or through gross negligence as it is obvious from the above documents, and thus such fatal flaws in the regulatory authorities are needed to be rigorously pursued.
The underestimation in the qPCR is a fundamental problem that directly relates not only to the regulated amount of the DNA contamination but also to human life, and thus to quantitatively eliminate it means a great advance in the DNA contamination problem.
Researchers focusing on the problem of DNA contamination have been aware of the underestimation caused by the qPCR but have not made any efforts to eliminate it. Researchers should not turn away from this underestimation problem and should take a stand toward eliminating the underestimation.
We intensively investigated a method to eliminate the underestimation based on the experimental results disclosed by McKernan and Buckhaults. The purpose of this post is to provide a first step for eliminating the underestimation in the qPCR.
The estimated value at present point shall now be disclosed hereinafter, but a certain correction value may be added depending on future experimental results, etc. However, we believe that a method for eliminating the underestimation has been established to some extent and disclose this here.
We also believe this post will provide deep insights and further perspectives on the DNA contamination problem to many researchers. We expect that excellent researchers will give further consideration to this.
This post updates and specifically explains the calculation method for the DNA contamination shown in the past post from mbi (ref. 7).
◆Why is the DNA contamination amount underestimated?
The underestimation of the DNA contamination amount is caused by a manufacturing process of the mRNA vaccine and a process of the qPCR (FIG. 1). Specifically, the underestimation is mainly caused from the following three steps.
(1) A cleaving step of the plasmid DNA with a DNase I treatment (Ref. 8, FIG. 2)
(2) A removal step of the plasmid DNA fragments smaller than a certain size with an ultrafiltration treatment
(3) A measurement step of the DNA fragments larger than a certain size with the qPCR
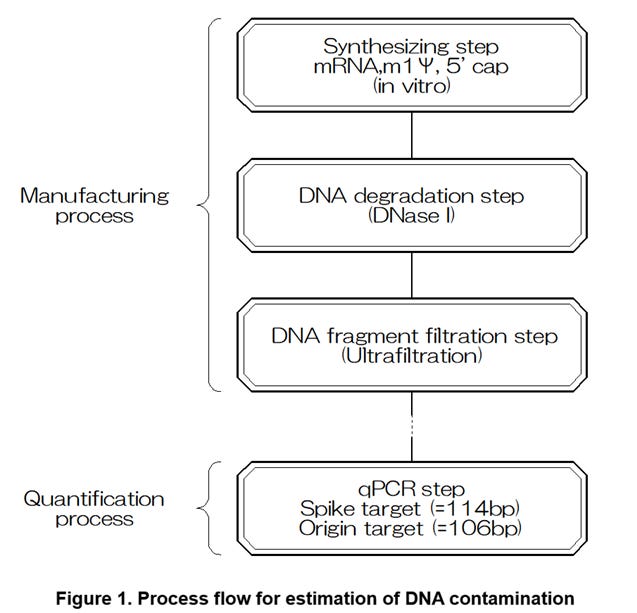
The steps (1) and (2) are included in the manufacturing process flow of the mRNA vaccine and are performed after a synthesizing step of the spike mRNA from the plasmid DNA. The step (3) is included in the process flow of the qPCR.
(1) The cleaving step
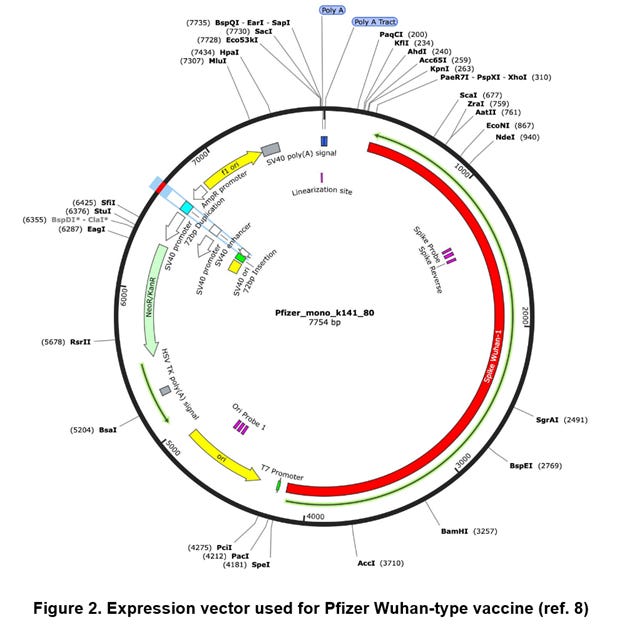
In this step, the plasmid DNA (=7754 [bp]) is cleaved into a predetermined average size with the DNase I treatment. That is, the qPCR targets are also cleaved into a certain probability, and therefore total numbers of intact qPCR targets (probes) are to be reduced. The qPCR targets are a part of a spike region (i.e., spike target=114 [bp]) and a part of an origin region (i.e., origin target=106 [bp]) (Ref. 8, FIG. 2).
(2) The ultrafiltration step
In this step, small DNA fragments not more than a predetermined size (=100 [bp] or less) among the DNA fragments cleaved in the cleaving step are to be removed. In this step, the intact qPCR targets (=114[bp], 106[bp]) are to be remained whereas the cleaved target fragments are to be removed. Note that all small DNA fragments are not necessarily removed in this step.
(3) The measurement step
In this step, the DNA contamination amount is measured based on the qPCR targets. Therefore, the DNA fragments smaller than the target size (114 [bp], 106 [bp]) cannot be detected in the qPCR.
Those steps (1) to (3) all lead to the underestimation of the DNA contamination amount. Hereinafter, a specific method for eliminating the underestimation and calculating the estimated amount of the DNA contamination shall now be explained.
◆The method for quantitatively eliminating the underestimation
According to the substack article of McKernan, it has been suggested that the spike region of the plasmid DNA forms a GC-rich mRNA/DNA hybrid with a modified spike mRNA (ref. 9). The spike region is a complementary DNA (i.e., cDNA) with respect to the spike mRNA, and is composed of the Crick strand ORF (open reading frame) region shown by the green line in the above FIG. 2 (ref. 8).
Furthermore, it also has been suggested that the spike region has a relatively high nuclease resistance whereas the origin region (i.e., vector region) has a relatively low nuclease resistance. This has been considered to be because the modified spike mRNA provides a protection to the spike region (ref. 7).
The spike mRNA has a structure in which uracil is replaced to m1Ψ, and this makes the melt temperature of the spike mRNA higher (ref. 10). In other words, a binding strength of the spike mRNA with respect to the spike region is increased.
In a case in which the estimated amount of the DNA contamination is calculated without considering a protection rate with the spike mRNA, an appropriate amount cannot be calculated from the spike region, and it is thus necessary to focus on the origin region. On the other hand, in a case in which the estimated amount of the DNA contamination is calculated with considering the protection rate with the spike mRNA, it is necessary to focus on the spiked region.
The estimated amount of the DNA contamination is to be determined through following Steps 1 to 14. The steps 1 to 7 show a determination method for the estimated amount of the DNA contamination without considering the protection rate with the spike mRNA, and the steps 8 to 14 show a determination method for the estimated amount of the DNA contamination with considering the protection rate with the spike mRNA.
(Step 1) Formularization of an average cleaved base number m
(Step 2) Calculation of an actual measurement amount No with the origin target
(Step 3) Calculation of an actual usage amount Nu of the plasmid DNA
(Step 4) Calculation of the average cleaved base number m
(Step 5) Formularization of a first estimated amount N1 of the DNA contamination after the ultrafiltration step
(Step 6) Calculation of a removal rate Rg with the ultrafiltration step
(Step 7) Calculation of the first estimated amount N1
(Step 8) Formularization of a second estimated amount N2 of the DNA contamination with considering a protection rate PR
(Step 9) Formularization of the protection rate PR
(Step 10) Calculation of a first DNA fragment number Nxs
(Step 11) Calculation of an existence probability Ps of the spike target
(Step 12) Calculation of the protection rate PR
(Step 13) Calculation of the second estimated amount N2
(Step 14) Another calculation method for the protection rate PR
Hereinafter, the Steps 1 to 14 shall now be explained in that order regarding the lot number EL9262 (Table 1). In addition, hereinafter, numerical values obtained from the calculation results using Microsoft Excel are shown. Therefore, it should be noted that if the numerical values shown below are used in calculations as they are, it will not necessarily match the numerical values shown in the Table.
Some considerations will be made after explained those steps 1 to 14.
◆(Step 1) Formularization of the average cleaved base number m
Where an average number z of cleaved portions can be randomly produced from the target DNA to be cleaved, the probability P that k-number of cleaved portions is to be produced from the target DNA follows the "Poisson distribution" (ref. 11). The formula of the Poisson distribution is as follows.
P=(e^(-z)×z^k)/k! …(1)
P: Probability
z: The average number of occurrences
k: The number of occurrences (k=0,1,2....)
e: Euler’s number (e=2.71828...)
!: Factorial function
When a base number of the target DNA is set as n [bp] and an average cleaved base number of the DNA fragments after the cleaving step is set as m [bp], the average number z of the cleaved portions to be produced from the target DNA is given by the following formula.
z=n/m …(2)
A probability that the target DNA exists in an intact state, that is, a probability P that zero number (k=0) of the cleaved portion is produced from the target DNA, is given by the following formula with the above formulas (1) and (2).
P=exp(-n/m) …(3)
The base number of the plasmid DNA is 7754 [bp] and the base number of the origin target in the qPCR is 106 [bp]. When the plasmid DNA is randomly cleaved so as to become the average cleaved base number m (=7754/m), the number of cleaved portions produced from the origin target becomes 106/m. From this, an existence probability Po that the origin target exists in an intact state is expressed from the above formula (3) as follows.
Po=exp(-106/m) …(4)
Most of the plasmid DNA fragments under 100 [bp] are selectively removed in the ultrafiltration step as described above. In other words, the intact origin target is not removed in this step since it has the size of 106 [bp]. Thus, an actual measurement amount No [g/dose] of the DNA contamination per dose calculated from a calibration curve is expressed following formula with an actual usage amount Nu [g/dose] of the plasmid DNA per dose used in the manufacturing process of the mRNA vaccine and the existence probability Po of the intact origin target. The actual usage amount Nu is also referred to as a templated usage amount.
No=Po×Nu …(5)
No=exp(-106/m)×Nu …(*5)
The following formula is obtained by solving the above formula (*5) about the average cleaved base number m.
m=-106/ln(No/Nu) …(6)
Hereinafter, the average cleaved base number m is calculated from the actual measurement amount No and the actual usage amount Nu of the plasmid DNA.
◆(Step 2) Calculation of the actual measurement amount No
The actual measurement amount No[g/dose] of the DNA contamination per dose when the origin target was used is expressed by the following formula.
No=(a×L×Mo×G)/NA …(7)
a: Dilution factor with pure water=10
L: Inoculation amount per dose=300 [ul/dose]
Mo: Molecule number [#/ul] of the plasmid DNA per 1 [ul]
G: Molecular weight of the plasmid DNA=3838872 [Da]
NA: Avogadro constant=6.02E+23 [/mol]
The molecule number Mo is unidentified among the above factors. The molecule number Mo can be calculated from the formula shown in the calibration curve of the origin target (FIG. 3) provided from Buckhaults (refs. 12 and 13). The formula shown in the calibration curve can be rewritten as the following formula with the molecule number Mo.
Cq=-3.790×log10Mo+43.043 …(8)

The molecule number Mo can be calculated by serving the Cq value as 20.6325 in the above formula (8) and solving it about the molecule number Mo.
Mo=8.19E+5 [#/ul] …(9)
From the above, the actual measurement amount No can be calculated as follows by substituting the above formula (9) into the above formula (7).
No=1.57E-8 [g/dose] …(*7)
◆(Step 3) Calculation of the actual usage amount Nu
The qPCRs according to the study of Buckhaults were performed to the vials manufactured through the Process 2 of Pfizer. The actual usage amount Nu [g/dose] of the plasmid DNA per dose can thereby be calculated from a usage amount Na [mg/ml] of the plasmid DNA and an yield amount Nb [mg/ml] of the spike mRNA which have been disclosed in the Tables on p.49 and p.59 of the Pfizer document (ref. 14).
The usage amounts Na are 0.09 to 0.110 [mg/ml] (Table 2A).
The yield amounts Nb are 2.19 to 2.21 [mg/ml] (Table 2B).
Here, the respective averages were calculated, the usage amount Na was set to 0.1 [mg/ml], and the yield amount Nb was set to 2.2 [mg/ml]. From those amounts, a usage rate Ru (i.e., template usage rate) of the plasmid DNA per 1 [ml] can be calculated from the following formula.
Ru=Na/Nb=0.0455 …(10)
A weight of the spike mRNA per dose is 30 [ug/dose]. Therefore, the actual usage amount Nu of the plasmid DNA per dose can be calculated as follows.
Nu=3E-5×Ru(=0.0455) …(11)
Nu=1.37E-6 [g/dose] …(*11)
An actual molecule number Nn [#/dose] of the plasmid DNA per dose before the ultrafiltration step is calculated from the following formula by using the actual usage amount Nu of the plasmid DNA, the molecular weight G of the plasmid DNA, and the Avogadro's constant NA.
Nn=Nu/G×NA …(12)
Nu=1.37E-6 [g/dose] …(*11)
G=3,838,872 [Da]
NA=6.02E+23 [/mol]
Nn=2.14E+11[#/dose] …(*12)
The actual molecule number Nn of the plasmid DNA will be used in later calculations.

◆(step 4) Calculation of the average cleaved base number m
The average cleaved base number m has been given by the formula (6) in the step 1.
m=-106/ln(No/Nu) …(6)
Furthermore, the actual measurement amount No has been calculated from the formula (*7) in the step 2, and the actual usage amount Nu has been calculated from the formula (*11) in the step 3.
No=1.57E-8 [g/dose] …(*7)
Nu=1.37E-6 [g/dose] …(*11)
From the above formulas, the average cleaved base number m can be calculated as follows.
m=-106/ln(1.57E-8/1.37E-6) …(*6)
m=23.73≃24 …(*6)
The existence probability Po of the intact origin target also can be calculated by substituting the average cleaved base number m (=24) into the formula (4) in the step 1.
Po=exp(-106/m)=exp(-106/24) …(4)
Po=0.0115 …(*4)
The existence probability Po of the origin target will be used in later calculations.
◆(step 5) Formularization of the first estimated amount N1
The first estimated amount N1 [g/dose] of the DNA contamination after the ultrafiltration step is a total weight of the residual DNA fragments which avoided the removal with the ultrafiltration step. Therefore, the first estimated amount N1 of the DNA contamination is calculated by multiplying the actual usage amount Nu [g/dose] of the plasmid DNA by a residual rate Rr [w/w] of a DNA fragment weight.
N1=Nu×Rr …(13)
Nu=1.37E-6 [g/dose] …(*11)
On the other hand, the residual rate Rr of DNA fragments is given by the following formula using the removal rate Rg [w/w] of the DNA fragment weight.
Rr=1-Rg …(14)
Hereinafter, the removal rate Rg with the ultrafiltration step shall now be calculated.
◆(step 6) Calculation of the removal rate Rg
To calculate the removal rate Rg of the DNA fragment weight, an existence probability Pn [#/#] of the DNA fragment number having an arbitrary base number s [bp] through the DNase I treatment and an existence probability Pg [w/w] of the DNA fragment weight are needed to be calculated.
Prior to the calculation of the removal rate Rg, here, figures in each of which the average cleaved base number m is set to 10 [bp], 50 [bp] and 200 [bp] are shown for better visual understanding (FIG. 4).

FIG. 4A shows a graph of an existence probability Pn [#/#] of the DNA fragment number, FIG. 4B shows a graph of a cumulative probability Pnc [#/#] of the DNA fragment number, FIG. 4C shows a graph of an existence probability Pg [w/w] of the DNA fragment weight, and FIG. 4D shows a graph of a cumulative probability Pgc [w/w] of the DNA fragment weight.
Those probability Pn, Pnc, Pg and Pgc were calculated by the following formulas based on the Poisson distribution, respectively. The distributions were calculated by varying the base number s [bp] in a range from 0 [bp] to 7754 [bp] in the following formulas.
Pn=exp(-s/m)-exp{-(s+1)/m}, [s=0 to 7754] …(15)
Pnc=ΣPn, [s=0 to 7754] …(16)
Pg=s×Pn×Ny, [s=0 to 7754] …(17)
Pgc=ΣPg, [s=0 to 7754] …(18)
m=10 [bp], 50 [bp], 200 [bp]
In the above formula (17), "Ny" is a DNA fragment number [#/dose] after the DNase I treatment. The DNA fragment number Ny can be calculated from the Poisson distribution as well. When a linear plasmid DNA is cut into the average cleaved base number m, a probability Py where a y-number of cutting sections is to be produced is expressed with the following formula using an average cut number u of the DNA fragments.
Py=(u^y/y!)×exp(-u) …(19)
u=7754/m …(20)
m=10 [bp], 50 [bp], 200 [bp]
When the y-number of cutting sections occurs in the linear plasmid DNA, the fragment numbers produced from the linear plasmid DNA become (y+1)-number. Therefore, a probability distribution Pyn of the DNA fragment number can be expressed with the following formula by using the actual molecule number Nn [#/dose] of the plasmid DNA per dose before the ultrafiltration step (see the formula (*12) in the step 3).
Pyn=(y+1)×(u^y/y!)×exp(-u)×Nn …(21)
Nn=2.14E+11 …(*12)
The DNA fragment number Ny is calculated from a total amount when the cutting section y is varied in the range from 0 [bp] to 7754 [bp] in the above formula (21).
Ny=ΣPyn, [y=0 to 7754] …(22)
Those calculation results are summarized in the above FIG. 4. As it is apparent from those graphs, in a case in which the average cleaved base number m is set to a relatively large value, the number and the weight of the small DNA fragments are decreased. On the other hand, in a case in which the average cleaved base number m is set to a relatively small value, the number and the weight of the small DNA fragments are increased. In particular, when the average cleaved base number m is set to a value smaller than 50 [bp], the number and the weight of the small DNA fragments are acutely increased.
From those graphs, it can be understood that the number and the weight of the small DNA fragments become extremely large in the target lot EL9262 this time since the average cleaved base number m is "24". Regarding the target lot EL9262, those probabilities Pn, Pnc, Pg, and Pgc can be calculated by substituting "m=24" in the above formulas (15) to (22).
The removal rate Rg [w/w] of the DNA fragment weight [g/dose] also can be calculated by substituting "m=24" in the above formulas (15) to (22). The average fragment number u shown in the above formula (20) is 327, and the DNA fragment number Ny shown in the above formula (22) is 7.00E+13 [#/dose].
Pn=exp(-s/m)-exp{-(s+1)/m},[s=0 to 7754] …(15)
Pnc=ΣPn,[s=0 to 7754] …(16)
Pg=s×Pn×Ny,[s=0 to 7754] …(17)
Pgc=ΣPg,[s=0 to 7754] …(18)
m=24 …(*6)
Nn=2.14E+11 …(*12)
u=327 …(*20)
Ny=7.00E+13[#/dose] …(*22)
In the Pfizer manufacturing process 2, the small DNA fragments were removed through ultrafiltration with the MWCO 300 [kDa] (Table 3, ref. 14, p.23). In this ultrafiltration step, almost all molecules of at least not more than 100 [bp] are to be removed as it is understood from the document regarding the MWCO (FIG. 5, ref. 15).

In a fragment analysis with the Oxford Nanopore Sequencing (ONT), a peak of the DNA fragments exists at approximately 100 [bp] (FIG. 6, refs. 12 and 13). It was therefore assumed here that the fragments of 100 [bp] or less were removed through the ultrafiltration step.

The removal rate Rg can be calculated by a rate of a total weight of the DNA fragments before and after the ultrafiltration step. Specifically, the removal rate Rg is a rate of a first total weight Pgc1 [g/dose] when the base number s is varied in a range from 0 [bp] to 7754 [bp] in the above formula (18) and a second total weight Pgc2 [g/dose] when the base number s is varied in a range from 0 [bp] to 100 [bp] in the above formula (18).
Rg=Pgc2/Pgc1 …(23)
Pgc1=ΣPg,[s=0 to 7754] …(24)
Pgc2=ΣPg,[s=0 to 100] …(25)
Rg=0.922 [w/w] …(*23)
On the other hand, a removal rate Rn [#/#] of the DNA fragment number Nn [#/dose] in the ultrafiltration step can be calculated by substituting "m=24" in the above formulas (15) to (22). Specifically, the removal rate Rn is a rate of a first total number Pnc1 [#/dose] when the base number s is varied a range from 0 [bp] to 7754 [bp] in the above formula (16), and a second total number Pnc2 [#/dose] when the base number s is varied a range from 0 [bp] to 100 [bp] in the above formula (16).
Rn=Pnc2/Pnc1 …(26)
Pnc1=ΣPn, [s=0 to 7754] …(27)
Pnc2=ΣPn, [s=0 to 100] …(28)
Rn=0.985 [#/#] …(*26)
In addition, since the DNA fragment number Ny [#/dose] after the DNase I treatment has been obtained from the above formula (*22), a rate Rf of the fragment number before and after the DNase I treatment can be calculated. The rate Rf of the fragment number is also another expression format of the average fragment number u. As shown below, the rate Rf of the fragment number is expressed by a rate of the DNA fragment number Ny [#/dose] after the DNase I treatment with respect to the actual molecule number Nn [#/dose] of the plasmid DNA before the DNase I treatment.
Rf=Ny/Nn …(29)
Nn=2.14E+11 [#/dose] …(*12)
Ny=7.00E+13 [#/dose] …(*22)
Rf=u=327 …(*29)
◆(step 7) Calculation of the first estimated amount N1
Since the removal rate Rg (=0.922 [w/w]) of the DNA fragment has been obtained from the formula (*23) in the step 6, the residual rate Rr [w/w] can be calculated by substituting the removal rate Rg into the formula (14) in the step 5.
Rr=1-Rg …(14)
Rg=0.922[w/w] …(*23)
Rr=0.0784[w/w] …(*14)
Furthermore, since the residual rate Rr of the DNA fragments has been obtained, the first estimated amount N1 of the DNA contamination after the ultrafiltration step can be calculated as follows by substituting the residual rate Rr into the formula (13) in the step 5.
N1=Nu×Rr …(13)
Nu=1.37E-6 [g/dose] …(*11)
Rr=0.0784 [w/w] …(*14)
N1=1.07E-7[g/dose] …(*13)
At this point, the first estimated amount N1 (=107[ng/dose]) of the DNA contamination is more than 10 times the regulation amount (=10[ng/dose]). Furthermore, since the weight of the spike mRNA per dose is 30 [ug/dose] (=3E-5 [g/dose]), an RNA/DNA rate of the spike mRNA and the DNA becomes 280. Therefore, the RNA/DNA rate is significantly below the regulation value (>3000).
◆(step 8) Formularization of the second estimated amount N2
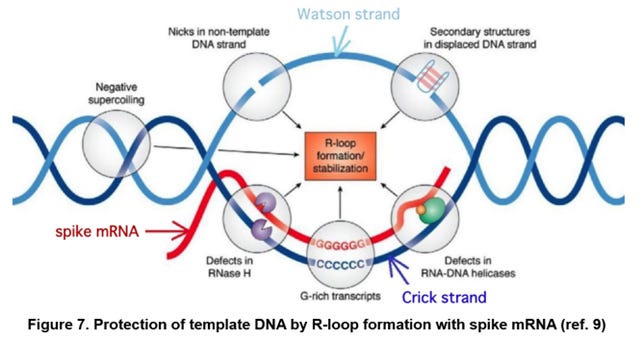
As described above, it has been suggested that the spike region (i.e., cDNA) of the plasmid DNA forms the GC-rich RNA/DNA hybrid with the modified spike mRNA (R-loop, FIG. 7, ref. 9). However, a protection rate PR with the spike mRNA has not been taken into consideration in the first estimated amount N1 of the DNA contamination.
Therefore, hereinafter, the protection rate PR is introduced, and the second estimated amount N2[g/dose] of DNA contamination is formulated. Hereinafter, it is assumed that both the Watson strand ORF and the Crick strand ORF constituting the R-Loop together with the spike mRNA are protected by the spike mRNA, and the second estimated amount N2 is calculated (FIG. 7).
Note that, in practice, it is possibly assumed that a case where only one of the Watson and Crick strand ORFs is protected by the spike mRNA, or a case where one or both of the Watson and Crick strand ORFs is or are partially protected by the spike mRNA.
The second estimated amount N2 of the DNA contamination is calculated from an unprotected side estimated amount N21 [g/dose] to be extracted from the plasmid DNAs which are not protected by the spike mRNAs, and a protected side estimated amount N22 [g/dose] to be extracted from the plasmid DNAs which are protected by the spike mRNAs.
N2=N21+N22 …(30)
The unprotected side estimated amount N21 is composed of a total amount of the DNA fragments which are not protected by the spike mRNA and are remained through the ultrafiltration step, in the actual usage amount Nu of the plasmid DNA. Therefore, the unprotected side estimated amount N21 is expressed by an integrated value of the actual usage amount Nu, the unprotected rate (1-PR) with the spike mRNA, and the residual rate Rr of the DNA fragments.
N21=Nu×(1-PR)×Rr …(31)
Nu=1.37E-6 [g/dose] …(*11)
Rr=0.0784 [w/w] …(*14)
PR=unidentified
On the other hand, the protected side estimated amount N22 is composed of a total amount of the DNA fragments which are protected by the spike mRNA and are remained through the ultrafiltration step, in the actual usage amount Nu of the plasmid DNA. The plasmid DNA has a region which is protected by the spike mRNA and a region which is not protected by the spike mRNA. The region protected by the spike mRNA is the complementary DNA region (i.e., cDNA) with respect to the spike mRNA in the plasmid DNA.
It is therefore necessary to know an occupation rate RD of a base number Ncd (=3822 [bp]) of the cDNA with respect to a base number Npl (=7754 [bp]) of the plasmid DNA. The occupation rate RD is given as follows.
RD=Ncd/Npl …(32)
RD=0.493 …(*32)
The protected side estimated amount N22 is composed of a total amount of a cDNA side estimated amount N22A [g/dose] of the DNA fragments to be extracted from the cDNAs side and an outside estimated amount N22B of the DNA fragments to be extracted from the regions outside the cDNAs.
N22=N22A+N22B …(33)
The cDNA side estimated amount N22A is composed of a total amount of the DNA fragments which are protected by the spike mRNAs and are remained through the ultrafiltration step, in the actual usage amount Nu [g/dose]. The residual rate of the cDNA protected by the spike mRNA at the ultrafiltration step is 1. Therefore, the cDNA side estimated amount N22A is expressed by an integrated value of the actual usage amount Nu, the protection rate PR with the spike mRNA, and the occupation rate RD of the cDNA.
N22A=Nu×PR×RD×1 …(34)
On the other hand, the outside estimated amount N22B is a total amount of the DNA fragments which are to be produced from regions outside the cDNAs protected by the spike mRNAs and are remained through the ultrafiltration step, in the actual usage amount Nu [g/dose]. Therefore, the outside estimated amount N22B is expressed by an integrated value of the actual usage amount Nu, the protection rate PR with the spike mRNA, an inoccupation rate (=1-RD) of the cDNA, and the residual rate Rr.
N22B=Nu×PR×(1-RD)×Rr …(35)
The second estimated amount N2 given by the above formula (30) can be rewritten as follows based on the above formulas (31) to (35).
N2=(Nu×(1-PR)×Rr)+(Nu×PR×RD×1)+(Nu×PR×(1-RD)×Rr) …(*30)
Nu=1.37E-6 [g/dose] …(*11)
Rr=0.0784 [w/w] …(*14)
RD=0.493 …(*32)
PR=unidentified
In the above formula (*30), the protection rate PR with the spike mRNA is still unidentified. Hereinafter, the protection rate PR shall now be calculated.
◆(step 9) Formularization of the protection rate PR
The protection rate PR can be calculated by using a first DNA fragment number Nxs [#/dose] when the spike target is used in the measurement step with the qPCR. The first DNA fragment number Nxs can be detected from both the intact spike target which is protected by the spike mRNA and the intact spike target which is not protected by the spike mRNA.
That is, the first DNA fragment number Nxs includes a first fragment number Ns1 [#/dose] detected due to the intact spike protected by the spike mRNA and a second fragment number Ns2 [#/dose] detected due to the intact spike target unprotected by the spike mRNA.
Nxs=Ns1+Ns2 …(36)
The spike targets protected by the spike mRNAs are all in intact state and are to be detected by the qPCR. Therefore, the first fragment number Ns1 can be calculated by multiplying the actual molecule number Nn [#/dose] per dose before the ultrafiltration step by the protection rate PR with the spike mRNA.
Ns1=Nn×PR …(37)
Nn=2.14E+11 [#/dose] …(*12)
On the other hand, the spike targets unprotected by the spike mRNAs are randomly cleaved through the DNase I treatment, and only the residual intact spike targets through the ultrafiltration step are to be detected by the qPCR. Therefore, the second fragment number Ns2 can be calculated by multiplying the actual molecule number Nn [#/dose] before the ultrafiltration step by the unprotection rate (=1-PR) with the spike mRNA, and an existence probability Ps of the intact spike target.
Ns2=Nn×(1-PR)×Ps …(38)
From the above, the first DNA fragment number Nxs given in the above formula (36) can be rewritten as follows by using the above formulas (37) and (38).
Nxs=Nn×PR+Nn×(1-PR)×Ps …(*36)
The above formula (*36) can be expressed as follows by solving it about the protection rate PR.
PR=(Nxs-Nn×Ps)/{Nn×(1-Ps)} …(39)
In the above formula (39), the first DNA fragment number Nxs when the spike target is used, and the existence probability Ps of the spike target are unidentified. Hereinafter, the first DNA fragment number Nxs and the existence probability Ps shall now be calculated.
◆(step 10) Calculation of the first DNA fragment number Nxs
The first DNA fragment number Nxs [#/dose] per dose when the spike target is used can be calculated by the following formula using an actual amount Ns [g/dose] of the DNA contamination when the spike target is used, the molecular weight G of the plasmid DNA, and the Avogadro's constant NA.
Nxs=Ns/G×NA …(40)
The actual amount Ns can be expressed by the following formula.
Ns=(a×L×Ms×G)/NA …(41)
a: Dilution factor with pure water=10
L: Inoculation amount per dose=300 [ul/dose]
Ms: Molecule number [#/ul] of the plasmid DNA per 1 [ul]
G: Molecular weight of the plasmid DNA=3838872 [Da]
NA: Avogadro constant=6.02E+23 [/mol]
The molecule number Ms is unidentified among the above factors. The molecule number Ms can be calculated from the formula shown in the above calibration curve of the spike target (FIG. 3) obtained from the experiment of Buckhaults (refs. 12 and 13). The formula shown in the calibration curve can be rewritten as the following formula using molecule number Ms.
Cq=-3.796×log10Ms+42.480 …(42)
The molecule number Ms can be calculated by serving the Cq value as 20.035 in the above formula (42) and solving it about the molecule number Ms.
Ms=8.18E+5[#/ul] …(43)
The actual amount Ns is calculated by substituting the above formula (43) into the above formula (41).
Ns=1.57E-8[g/dose] …(*41)
From the above formulas, the first DNA fragment number Nxs can be calculated by substituting the above formula (*41) into the above formula (40).
Nxs=2.45E+9[#/dose] …(*40)
◆(step 11) Calculation of the existence probability Ps
The existence probability Ps of the spike target is calculated by the following formula as with the formula (3) of the existence probability Po of the origin target in the step 1. The average cleaved base number m has been already obtained in the formula (*6) in the step 4.
Ps=exp(-114/m) …(44)
m=24 …(*6)
Ps=0.0082 …(*44)
◆(step 12) Calculation of the protection rate PR
The protection rate PR has been given in the formula (39) in the step 9. Furthermore, the actual molecule number Nn has been already obtained from the formula (*12) in the step 3, the first DNA fragment number Nxs has been already obtained from the formula (*40) in the step 10, and the existence probability Ps has been already obtained from the formula (*44) in the step 11. The protection rate PR can thereby be calculated as follows.
PR=(Nxs-Nn×Ps)/{Nn×(1-Ps)} …(39)
Nn=2.14E+11 [#/dose] …(*12)
Nxs=2.45E+9 [#/dose] …(*40)
Ps=0.0082 …(*44)
PR=3.30E-3 [#/#] …(*39)
◆(step 13) Calculation of the second estimated amount N2
The second estimated amount N2 has been given in the formula (*30) in the step 8. Furthermore, the protection rate PR has been already obtained from the formula (*39) in the step 12. The second estimated amount N2 can thereby be calculated as follows.
N2=(Nu×(1-PR)×Rr)+(Nu×PR×RD×1)+(Nu×PR×(1-RD)×Rr) …(*30)
Nn=2.14E+11 [#/dose] …(*12)
PR=3.30E-3 [#/#] …(*39)
Nxs=2.45E+9 [#/dose] …(*40)
Ps=0.0082 …(*44)
N2=1.09E-7 [g/dose] …(*30)
As described above, the second estimated amount N2 (=107 [ng/dose]) of the DNA contamination when the protection rate PR is taken into consideration is more than 10 times the regulation amount (=10 [ng/dose]). Furthermore, since the weight of the spike mRNA per dose is 30 [ug/dose] (=3E-5 [g/dose]), an RNA/DNA rate of the mRNA and the DNA becomes 275. Therefore, the RNA/DNA rate is significantly below the regulation value (>3000).
◆(step 14) Another calculation method for the protection rate PR
The actual molecule number Nn [#/dose] of the plasmid DNA before the ultrafiltration step (see the formula (*12) in the step 3) also can be given by the following formula using a second DNA fragment number Nxo [#/dose] per dose when the origin target is used, and the existence probability Po of the origin target (see the formula (*4) in the step 4). The second DNA fragment number Nxo is an amount assuming that the origin target is not protected by the spike mRNA.
Nn=Nxo/Po …(45)
The protection rate PR has been given by the formula (39) in the step 9 can be rewritten as follows by substituting the above formula (45).
PR=(Nxs×Po-Nxo×Ps)/(Nxo-Nxo×Ps) …(46)
PR=(Nxs-Nn×Ps)/{Nn×(1-Ps)} …(39)
Nn=Nxo/Po …(45)
That is, the above formula (46) is expressed by the first DNA fragment number Nxs [#/dose] when the spike target is used, the second DNA fragment number Nxo [#/dose] when the origin target is used, the existence probability Ps of the spike target, and the existence probability Po of the origin target.
The second DNA fragment number Nxo[#/dose] when the origin target is used can be calculated from the following formula using an actual measurement amount No [g/dose] of the DNA contamination per dose when the origin target is used (see the formula (*7) in the step. 2).
Nxo=No/G×NA …(47)
No=1.57E-8 [g/dose] …(*7)
G=3838872 [Da]
NA=6.02E+23 [/mol]
Nxo=2.46E+9 [#/dose] …(*47)
From the above formulas, the protection rate PR can be calculated as follows.
PR=3.30E-3 [#/#] …(*45)
Po=0.0115 …(*4)
Ps=0.0082 …(*44)
Nxo=2.46E+9 [#/dose] …(*47)
Nxs=2.45E+9 [#/dose] …(*40)
◆CONCLUSION
The above is the calculation method for the lot number EL9262. The first estimated amount N1 of the DNA contamination without considering the protection rate PR was 1.07E-7 [g/dose], and the second estimated amount N2 of the DNA contamination with considering the protection rate PR was 1.09E-7 [g/dose].
The first estimated amount N1 and the second estimated amount N2 were both more than 10 times the regulation amount (=10 [ng/dose]). In addition, the RNA/DNA rates were 280 and 275, respectively, and they were significantly lower than the regulation value (>3000). The protection rate with the R-Loop was 3.30E-3 [#/#].
Similar calculations were applied to the lot number EL9264. In this case, the first estimated amount N1 of the DNA contamination without considering the protection rate PR was 7.78E-8 [g/dose], and the second estimated amount N2 of the DNA contamination with considering the protection rate PR was 7.94E-8 [g/dose].
The first estimated amount N1 and the second estimated amount N2 were both more than 7 times the regulation value (=10 [ng/dose]). In addition, the RNA/DNA rates were 385 and 378, respectively, and they were significantly lower than the regulation amount (>3000). The protection rate with the R-Loop was 2.46E-3 [#/#].
◆The study of McKernan
The above calculation method for eliminating the underestimation in the qPCR was also applied to the two Pfizer vials (bivalent type and Wuhan-type) and one Moderna vial disclosed in the preprint of McKernan (ref. 1). These results are shown in Table 4.

Regarding the Moderna vial, the usage amount Na [mg/ml] of the plasmid DNA and the yield amount Nb [mg/ml] of the mRNA have not been disclosed by Moderna, and thus the actual usage amount Nu and the usage rate Ru according to the step 3 are unidentified and the average cleaved base number m cannot be calculated.
Therefore, calculations here were performed assuming that the template usage rate was 0.0455, which is the same value as that of the Pfizer. It should be noted that calculation results to the Moderna vials are analogous results when the template usage rate of Pfizer is adopted.
◆The results without considering the protection rate PR
The estimated amount of the DNA contamination of the Pfizer bivalent type vial was 28.7 [ng/dose] (=2.87E-08 [g/dose]), and the estimated amount of the DNA contamination of the Pfizer Wuhan-type vial was 81.9 [ng/dose] (=8.19E-08 [g/dose]), and the estimated amount of the DNA contamination of the Moderna vial was 68.4 [ng/dose] (=6.84E-08 [g/dose]). Therefore, all of those amounts significantly exceeded the regulation amount (=10 [ng/dose]).
In addition, the RNA/DNA rate of the Pfizer bivalent type vial was 1044, the RNA/DNA rate of the Pfizer Wuhan-type vial was 366, and the RNA/DNA rate of the Moderna vial was 731. Therefore, all of those amounts were significantly below the regulation amount (>3000).
◆The results with considering the protection rate PR
The estimated amount of the DNA contamination of the Pfizer bivalent type vial was 58.7 [ng/dose] (=5.87E-08 [g/dose]), and the estimated amount of the DNA contamination of the Pfizer Wuhan-type vial was 93.0 [ng/dose] (=9.30E-08 [g/dose]), and the estimated amount of the DNA contamination of the Moderna vial was 116 [ng/dose] (=1.16E-07 [g/dose]). Therefore, all of those amounts significantly exceeded the regulation amount (=10 [ng/dose]).
In addition, the RNA/DNA rate of the Pfizer bivalent type vial was 511, the RNA/DNA rate of the Pfizer Wuhan-type vial was 323, and the RNA/DNA rate of the Moderna vial was 432. Therefore, all of those amounts were significantly below the regulation amount (>3000).
◆CONCLUSION
As described above, it is understood from the study of McKernan that there is a possibility that not only the Pfizer vials but also the Moderna vials may significantly exceed the regulation amount (=10 [ng/dose]).
◆The DNA fragments having 630 [bp] or more
McKernan has reported in his substack article that the fragment number of 630 [bp] or more was 1.6 billion (=1.6E+9 [#/dose]) per dose (ref. 16).
A calculated existence probability of the DNA fragment number of 630 [bp] or more at the unprotected region outside the R-Loop in the plasmid DNA was approximately 10E-14 [#/dose], and it was found that the DNA fragments of 630[bp] or more were hardly produced from this region. That is, it is considered that the DNA fragments of 630 [bp] or more tend to be produced from the region protected by the R-Loop.
When the protection rate PR [#/#] was multiplied by the actual molecule number Nn of the DNA fragments per dose before the ultrafiltration step, the DNA fragment number of the Pfizer bivalent type vial was 9.76E+9 [#/dose], the DNA fragment number of the Pfizer Wuhan-type vial was 3.74E+9 [#/dose], and the DNA fragment number of the Moderna vial was 1.36E+10 [#/dose].
When similar calculations were applied to the study provided from Buckhaults, the DNA fragment numbers of 630[bp] or more were 7.07E+8[#/dose] and 5.27E+8[#/dose] (Table 1). Those numerical values were slightly lower compared with the study (=1.6E+9 [#/dose]) provided from McKernan.
In a case in which the ONT is used for the detection, a concentration of magnetic beads is usually lowered to remove the small DNA fragments when the DNA fragments are purified by an Ampure (ref. 17). In this connection, Buckhaults deliberately increased the concentration of the magnetic beads by a factor of 2.5 to eliminate this bias inclined toward the underestimation.
However, even in that case, as a result of the small DNA fragments passing through the magnetic beads, the small DNA fragments may be underestimated, and the rate of the large DNA fragments may become relatively high. Therefore, a number of the large DNA fragments with respect to the total number of the DNA fragments may be overestimated compared to an actual number. In other words, the DNA fragment number provided by McKernan may be slightly underestimated, and at the same time, the proportion of DNA fragments of 630 [bp] or more may be slightly overestimated.
Where the fragment distribution when the DNA fragments below 100 [bp] are removed by the Poisson distribution simulation and the result of the DNA fragment distribution analysis by the ONT (refs. 11, 12) are compared, the small molecules in a range from 100 [bp] to 190 [bp] are to be fitted with approximately m=90 [bp], but as the molecule number increases, it can be fitted with a value closer to 24 [bp] (FIG. 6C). This supports the idea that the measurement of the fragment distribution with the ONT is to be biased against the removal of small molecules.
From the above, accuracy may be increased by designing an assay where the concentration of the magnetic beads is increased and by measuring both the small DNA fragment numbers and the large DNA fragment numbers of 630 [bp] or more.
◆The study of Speicher
A further preprint studying on the Pfizer vials and the Moderna vials was filed on October 19, 2023, by Speicher in Canada (ref. 18). The above calculation method for eliminating the underestimation in the qPCR was also applied to all vials disclosed in the Table 2 of this preprint. The results are attached here (Table 5).

The Table 5 shows the DNA fragment amounts calculated from the calibration curve. However those amounts have not been taken into consideration the existence probability and the removal rate with the ultrafiltration step. Therefore, the actual measurement amount No was calculated from those amounts, and the average cleaved base number m was estimated thereafter.
m=-106/ln(No/Nu) …(6)
No: Actual amount in preprint Table 2
Nu: 1.37E-9 [g/dose] (Pfizer)
Nu: 2.28E-9 [g/dose] (Moderna)
The calculation method for the Moderna vials is similar to that described above. That is, the calculation was performed by serving the template usage rate as 0.00455, since the actual usage amount Nu (i.e., template usage rate) is unidentified as mentioned above. Therefore, it should be noted that calculation results to the Moderna vials are analogous results when the template usage rate of Pfizer is adopted.
◆The Pfizer vials
The estimated amounts of the DNA contamination in the 6 vials of the 8 vials exceeded the regulation amount (=10 [ng/dose]). The estimated amounts of two vials (FX4343a and FX4343b) were below the regulation amount.
The average cleaved base numbers m were 12 [bp] to 18 [bp]. The protection rates PR [w/w] with the R-Loop were zero percent to several percent. Some vials have a negative protection rate PR, but this means they are at the limit of detection.
When the numbers of the long DNA fragments of 630[bp] or more were calculated, some vials had 1E+7 [#/dose] or more whereas some vials were at the detection limit.
Therefore, it can be said that the Pfizer vials have a variation problem among lots and a variation problem due to the type of vaccine (that is, the type of the spike mRNA).
◆The Moderna vials
On the other hand, in the case of Moderna, the estimated amounts of the DNA contamination were all less than 10 [ng/dose] and tended to be lower than those of Pfizer, but some vials had relatively large DNA contamination amounts (=from 3 [ng/dose] to 5 [ng/dose]).
The average cleaved base numbers m were 9 [bp] to 11 [bp], and those were smaller (approximately half) than those of Pfizer. In other words, the DNase treatment of Moderna is superior to that of Pfizer. The protection rates PR [w/w] with the R-Loop were several percent to several tens of percent, and thus the protection rates PR of Moderna tended to be higher than those of Pfizer.
The DNA contamination amounts of Moderna were all relatively low, but the numbers of the relatively large DNA fragments of 630 [bp] or more were 1E+7 [#/dose] or more in all vials. That is, the risk of the presence of the relatively large DNA fragments (that is, the physical risk caused by the large DNA fragments) is higher for Moderna than for Pfizer.
Where the three BA.1 bivalent vials were excluded as outliers, a strong positive correlation (R=0.8485) was observed between the DNA contamination amounts and the large DNA fragment numbers of 630 [bp] or more (FIG. 8, ref. 18).

In the case of Moderna, since the DNA contamination amount is relatively low whereas the protection rate PR is relatively high, it is highly likely that the DNase treatment method is different from that of Pfizer. For example, the Moderna patent US10077439B2 discloses the new method for removing residual DNA (ref. 2), and this patent is one of arguable references.
As described above, it can be said that the Moderna vials also have a variation problem among lots and a variation problem due to the type of vaccine (that is, the type of the spike mRNA).
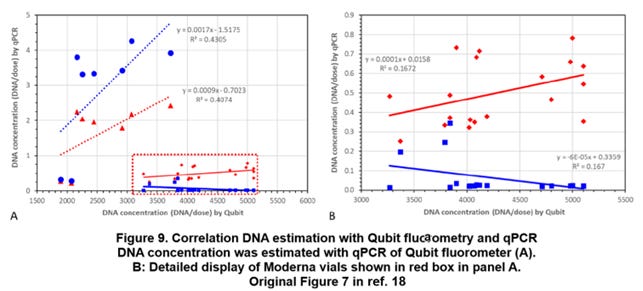
◆Further considerations for the Moderna vials
The Moderna vials have many uncertain points. For example, in the case of Moderna, the average cleaved base numbers m are around 10 [bp], but the Qubit results has shown relatively large amounts (FIG. 9). If the average cleaved base numbers m become around 10 [bp], most of the DNA fragments should be removed by the ultrafiltration step.
Even if all the spike regions as the cDNAs consisting of the R-loops remain, nearly half of the plasmid DNAs should be removed, but, in this case, more than 10 [ug] of the plasmid DNAs are used to produce the spike mRNAs, and thus this becomes an unrealistic calculation.
As it has been disclosed in the preprint of Speicher (ref. 18), in the case of Moderna, three vials AS0467Da, AS0467Db and AS0467Dc have irregular values. Referring to the protection rates PR of those vials, it is understood that each of the protection rates PR has one order of magnitude lower than those of other vials.
In other words, the measured amounts with the Qubit become relatively high in the vials having relatively high protection rates PR, and as a result, plots are shifted in parallel to the right direction in the graph (FIG. 9) showing the correlation between the measured amounts with the Qubit and the measured amounts with qPCR.
On the other hand, in the case of the spike target, the increase due to the protection with the spike mRNA can be detected by the qPCR. Therefore, in this case, it can be inferred that the irregularity caused by the protection rate PR is corrected as a result of the plots shifting upwards compared to the case of the origin target.
In the Qubit quantification step, Accu-Green was used as a dye, but it should be noted that the Accu-Green detects dsDNA by intercalating into double strands as with the SYBR Green. In this case, higher-order structures may account for a relatively large proportion in an absorbance with the Qubit, and as a result, the Qubit measured value may be overestimated.
mRNA/DNA hybrid, mRNA/mRNA hybrid, DNA/DNA hybrid and the like are exemplified as the higher-order structure. In fact, an RNase A treatment lowers the Qubit measured values in the Pfizer Wuhan-type vials by one order of magnitude from about 2 [ug/dose] to about 200 [ng/dose] (ref. 18). This suggests that the Qubit overestimates the amount of the DNA contamination because it detects the duplexes such as the mRNA/DNA hybrid and the mRNA/mRNA hybrid.
In particular, in a vial passed its storage period, the binding force of the mRNA/DNA hybrid, the mRNA/mRNA hybrid and/or the DNA/DNA hybrid may become stronger, and the possibility that many double strands are subsequently formed in the vial cannot be ruled out. The influence of the spike mRNA modified by m1Ψ can also be considered to the one cause of this.
Regarding the Pfizer Wuhan-type vial, the Qubit measured amount through the RNase A treatment is about 200 [ng/dose], and this is approximately twice the amount of 93 [ng/dose] theoretically estimated from the result of McKernan (Table 4). Regarding this lot, many undesirable side reactions have been reported, and it is suggested that a relatively large amount of DNA fragments exist in this vial. Therefore, the theoretical correction made here is considered to be almost appropriate on the basis the events occurred in this lot.
Those results suggest that running the Qubit on samples after the RNase treatment will yield more accurate data and deepen understanding. In addition, it is considered essential for the quantitative method to obtain the calibration curve by using the spike mRNA modified by m1Ψ.
The Qubit can selectively measure ssDNA, dsDNA, ssRNA, dsRNA, etc. It is therefore considered that accurate measurement of the DNA fragment amount will be possible by appropriately combining those targets and performing appropriate subtraction of the measured values.
◆Consideration for the Moderna patent
The disclosure of Moderna patent US10077439B2 (ref. 2) was further considered. This patent specification discloses a method for purifying the plasmid DNAs using a solid support. The solid support is commonly known as an affinity column. The calculation was made to estimate the DNA contamination amount assuming that the affinity column as the solid support was used in the purifying step based on the patent disclosure. The calculated results are shown in Tables 6 and 7 attached here. The experimental results provided from McKernan and Speicher were used here.
When the plasmid DNAs are purified by the affinity column, the plasmid DNAs are not degraded. Therefore, the existence probability of the plasmid DNAs become 1 and the removal rate of the plasmid DNAs become 0. Thus, the numbers of the spike targets and the origin targets in the DNA contamination should be equal each other.
However, in the experimental results provided from McKernan and Speicher, the numbers of the plasmid DNAs calculated from the Cq values and the uncorrected weights [ng/dose] by using the spike target and the origin target are significantly different each other. Therefore, it appears that the DNase treatment and the ultrafiltration step are actually implemented in the Moderna vials.
From these facts, a case where the purification process with the affinity column was combined with the DNase treatment and the ultrafiltration step was assumed here.
As a prerequisite, it was assumed that the plasmid DNA amount was reduced to one percent by the affinity column. In this case, the plasmid DNA amount before the DNase treatment was 23 [ng] and the DNA/RNA rate before the DNase treatment was 0.0005.
If the average cleaved base numbers m were calculated in this prerequisite, they became 96.44 [bp] for the experimental result of McKernan (Table 6), and 14 [bp] to 15 [bp] (outlier 22 to 25 [bp]) for the experimental result of Speicher (Table 7). In other words, when compared with the calculation method described above (Tables 4 and 5), the DNase treatment and the removal rate became insufficient since the average cleaved base numbers m became larger (Tables 6 and 7).

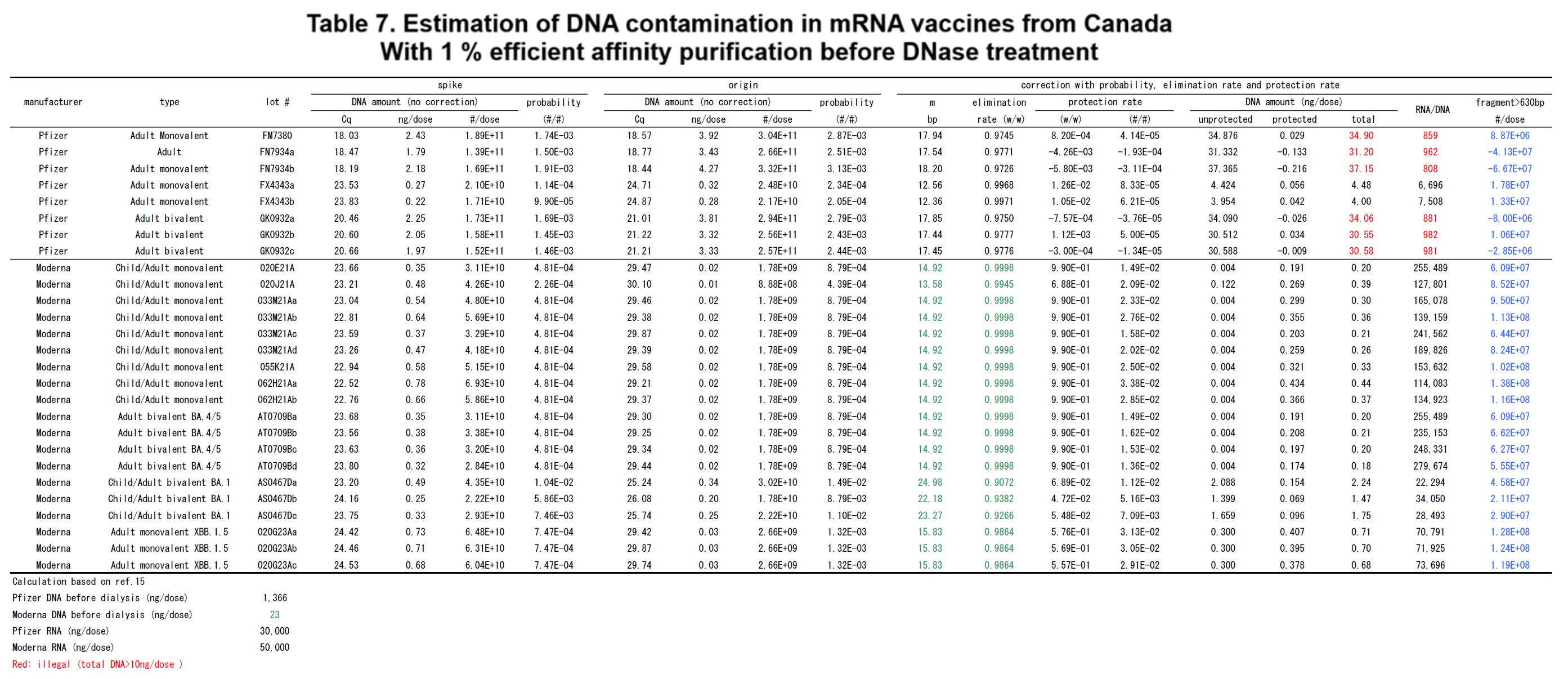
Specifically, in the case of the result provided from McKernan, the protection rate in the number [#/#] became 5.35 [#/#] and the protection rate in the weight [w/w] became 3.02 [w/w]. From these results, since the protection rate cannot be greater than 1, it is understood that almost all cDNAs are protected by the spike mRNAs here. On the other hand, in the case of the provided from Speicher, the protection rate [#/#] became 1/100 order from 1/10000 order.
Since the DNA purification with the affinity column can be performed under mild conditions for a long time, it can be expected that the probability of the R-loop formation increases, unlike when the strong DNase I treatment is performed immediately after the synthesizing step (FIG. 1). Even in this case, significant changes in the DNA contamination amounts are not observed.
In the case of Moderna, the amount of the plasmid DNAs before the DNase I treatment may be smaller on the first place rather than the DNase I treatment is superior (that is, the average cleaved base number m is smaller). This is a new interesting consideration.
Although it is not obvious whether Moderna is actually implementing the patented invention itself, the possibility that the affinity column is used in the Moderna manufacturing process should not be ruled out. For this issue, further research on Moderna vials and disclosure of manufacturing method of Moderna will be awaited.
The above are the calculation method for quantitatively eliminating the qPCR underestimation and the considerations obtained based on it.
Lastly, we believe this post will provide many researchers with deep insight and further perspective on the DNA contamination problem. We also hope that further efforts will be made to eliminate the underestimation of the qPCR from this post.
Thank you.
◆Reference lists
https://image-ppubs.uspto.gov/dirsearch-public/print/downloadPdf/10077439
https://image-ppubs.uspto.gov/dirsearch-public/print/downloadPdf/20130259924
https://image-ppubs.uspto.gov/dirsearch-public/print/downloadPdf/20190240317
https://www.pmda.go.jp/drugs/2021/P20210519003/400256000_30300AMX00266_B100_4.pdf
https://anandamide.substack.com/p/sequencing-the-pfizer-monovalent
https://anandamide.substack.com/p/dna-rna-hybrids-r-loops-and-nuclease
https://drive.google.com/drive/folders/1ZBj3N5P_yZ2QRgCV2pv3D4SEErbla6oL
https://anandamide.substack.com/p/spider-webs-in-the-pfizer-closet
https://anandamide.substack.com/p/dna-fragments-detected-in-monovalent
Deviser: mbi
Assistant: CultiVate 2
Author: Patent SUN and mbi
Translation (JP to EN): Patent SUN